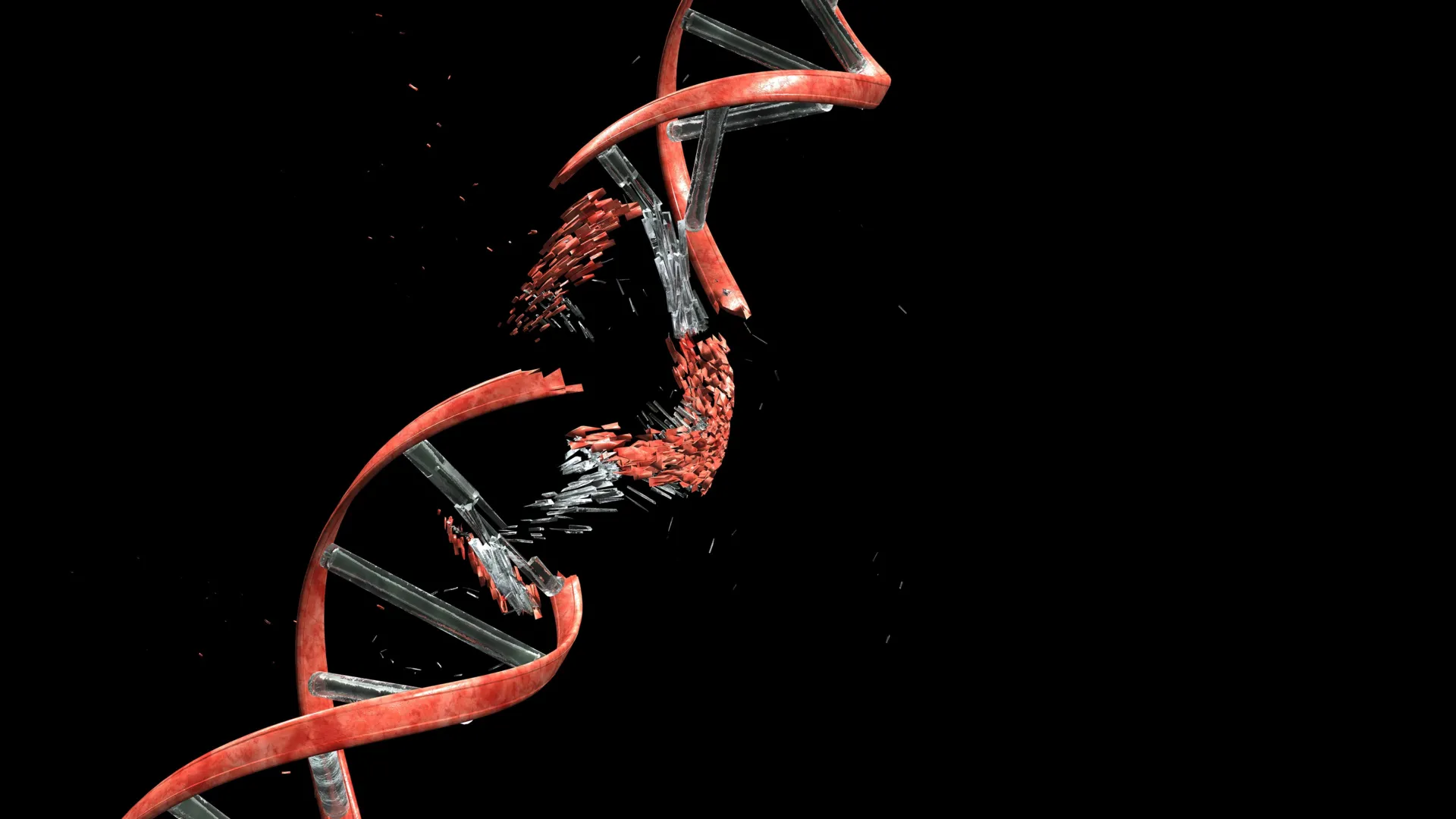
As the intricate architecture of the brain takes shape, a critical and arduous journey unfolds for newly formed neurons. These nascent cells, destined to become the fundamental units of our cognitive and sensory processing, must navigate a densely packed environment within the developing cerebral cortex. This perilous passage, characterized by squeezing through narrow interstitial spaces and circumventing neighboring cells, was recently found to inflict a surprising toll: significant DNA damage. A groundbreaking study, published in the esteemed journal Nature, has unveiled that these migratory neurons routinely sustain double-strand breaks in their DNA, a severe form of genetic injury where both strands of the DNA double helix are severed.
While double-strand breaks are widely recognized as potent instigators of mutations, cellular dysfunction, and even programmed cell death, this research from Kyoto University’s Institute for Integrated Cell-Material Sciences (WPI-iCeMS) and its collaborators reveals a paradoxical reality. In the context of healthy brain development, these breaks are not a harbinger of doom but rather a transient, albeit significant, consequence of cellular migration. Crucially, the developing brain possesses a remarkable capacity to rapidly and efficiently repair this damage, averting long-term deleterious effects and ensuring the integrity of the neuronal communication network.
Professor Mineko Kengaku, the lead researcher from WPI-iCeMS, articulated the study’s core finding: "The developing brain appears to have evolved to tolerate and repair the neuronal damage efficiently. But understanding the limits of that tolerance—and what happens when repair is incomplete—brings us closer to understanding a range of neurological conditions." This sentiment underscores the study’s profound implications, extending beyond fundamental developmental biology to offer potential insights into the origins of various neurological disorders.
The Mechanical Stress of Neuronal Migration
To meticulously investigate the origins of this DNA damage, the research team ingeniously recreated the formidable physical challenges encountered by migrating neurons. They employed sophisticated microfluidic devices, fabricating tiny channels designed to precisely mimic the constricted environments characteristic of growing brain tissue. By guiding cultured neurons through these microchannels, researchers could directly observe and analyze the cellular responses to mechanical stress.
Employing cutting-edge fluorescence microscopy and specialized DNA-labeling techniques, the scientists were able to visualize the formation of double-strand breaks as neurons actively propelled themselves through the confined spaces. A critical observation was that once the neurons successfully emerged from these restrictive channels, the DNA damage began to recede. The study meticulously documented that the vast majority of these double-strand breaks were repaired within a 24-hour period, a testament to the robust endogenous repair mechanisms present in these developing cells. Following successful repair, the neurons continued to exhibit normal functionality, seamlessly integrating into the developing neural circuits.
Unraveling the Molecular Culprit: Topoisomerase IIα
The research pinpointed a key molecular player in this process: Topoisomerase IIα (Topo IIα). This enzyme is a critical component of cellular machinery, normally tasked with managing DNA topology and mitigating the torsional stress that accumulates during essential cellular processes like DNA replication and transcription. Under physiological conditions, Topo IIα functions by transiently cleaving one or both strands of the DNA double helix to relieve supercoiling and tension, subsequently rejoining the broken ends. This mechanism can be analogized to carefully cutting and then re-splicing a twisted electrical cable to untangle it.
However, the study revealed that when neurons are subjected to intense mechanical stress, such as that encountered during their arduous migration through crowded brain tissue, Topo IIα can become "trapped" in a transient cleavage state. This mechanical perturbation can impede the enzyme’s ability to re-ligate the DNA strands, leaving behind unrepaired double-strand breaks. The cell then mobilizes a primary DNA repair pathway known as non-homologous end joining (NHEJ) to meticulously re-establish the broken DNA strands.
Differential Resilience: Neurons vs. Cancer Cells
A striking aspect of the study involved a comparative analysis of neuronal DNA damage with that observed in other cell types, specifically cancer cells, navigating similar microchannels. The researchers noted a significant divergence in the nature and consequences of DNA damage. In cancer cells, DNA damage often manifests more randomly, potentially disrupting critical cellular processes, leading to genomic instability, and ultimately triggering apoptosis (programmed cell death). This susceptibility to DNA damage contributes to the rationale behind certain cancer therapies that target DNA repair pathways.
In stark contrast, the DNA breaks observed in migratory neurons exhibited a distinct pattern. The researchers found that these breaks were predominantly localized to genomic regions that are not actively transcribed or involved in the expression of essential genes. This strategic localization is hypothesized to be a key factor in neuronal survival. By largely sparing the actively functioning, critical genes, the neurons could maintain their vital cellular functions and developmental trajectory despite the temporary presence of DNA double-strand breaks. This suggests a finely tuned evolutionary adaptation where the inherent risks of migration are mitigated by concentrating damage in less critical genomic areas.
The Peril of Incomplete Repair: Insights from Genetically Engineered Mice
To elucidate the functional consequences of compromised DNA repair during neuronal migration, the researchers ingeniously engineered a model organism. They developed mice with a genetic deficiency in Ligase 4, a crucial enzyme essential for the proper functioning of the NHEJ pathway, the primary mechanism for repairing double-strand breaks. Specifically, these mice were engineered to lack Ligase 4 in their newly formed cerebellar neurons.
Remarkably, these genetically modified mice initially developed without any apparent overt abnormalities. Their early life stages appeared normal. However, as they matured into adulthood, a subtle yet progressively worsening neurological phenotype began to emerge. These mice started exhibiting mild balance impairments, a symptom that gradually intensified over time. This phenotype bears a striking resemblance to certain human neurological disorders characterized by genome instability and cerebellar dysfunction, lending significant weight to the study’s findings regarding the critical role of DNA repair in maintaining long-term neurological health.
The cerebellum, responsible for motor control, coordination, and balance, is a highly complex structure where precise neuronal connectivity is paramount. The observed balance problems in the Ligase 4-deficient mice strongly suggest that even minor, cumulative DNA damage that escapes repair can ultimately compromise the intricate circuitry and functional integrity of this vital brain region. This finding provides a tangible link between the molecular mechanisms of DNA repair during development and the emergence of complex neurological symptoms later in life.
Broader Implications for Brain Diversity and Disease Etiology
The comprehensive findings of this study suggest that DNA breakage and repair processes may play a more profound and integral role in brain biology than previously understood. The research opens new avenues for investigation, prompting scientists to explore whether these early-life DNA alterations contribute to the inherent diversity observed among individual neurons. Each neuron, originating from the same genetic blueprint, might accumulate unique "scars" from its migratory journey, leading to subtle functional variations.
Furthermore, the study raises compelling questions about the potential contribution of these DNA damage and repair dynamics to the etiology of neurodevelopmental and neurodegenerative diseases. Could subtle, unrepaired DNA lesions during critical developmental windows predispose individuals to conditions like autism spectrum disorder, schizophrenia, or even later-onset neurodegenerative diseases such as Alzheimer’s or Parkinson’s? The research provides a compelling framework for exploring these critical questions.
Professor Kengaku elaborated on this paradigm shift: "It shifts how we think about the neuronal genome. All neurons originate from the same DNA, but DNA damage and repair can introduce small genetic differences between individual neurons through a small mechanical journey. Some of that history may be written into the genome itself." This perspective suggests that the genome is not a static entity but a dynamic landscape shaped by the very physical experiences of the cell, imprinting a unique history within the DNA itself.
The collaborative nature of this research, involving leading institutions such as Kyoto University, the University of Tokyo, the University of Osaka, the National University of Singapore, and the Tokyo Metropolitan Institute of Medical Science, highlights the global effort to unravel the complexities of brain development and function. The study’s rigorous methodology, innovative experimental design, and significant findings mark a pivotal moment in our understanding of how the brain builds itself and the unexpected challenges its constituent cells must overcome. Future research will undoubtedly build upon this foundation, seeking to precisely map the genomic landscape of DNA damage and repair, quantify its impact on neuronal function and connectivity, and ultimately translate these fundamental insights into novel strategies for preventing and treating debilitating neurological conditions. The intricate dance of neuronal migration, it appears, is not just a physical feat but a testament to the brain’s remarkable capacity for resilience, even in the face of inherent genetic vulnerability.
